
Variant Creutzfeldt-Jakob Disease (vCJD)
Variant Creutzfeldt-Jakob disease (vCJD) is a prion disease that was first described in 1996 in the United Kingdom. There is now strong scientific evidence that the agent responsible for the outbreak of prion disease in cows, bovine spongiform encephalopathy (BSE or ‘mad cow’ disease), is the same agent responsible for the outbreak of vCJD in humans.
Diagnostic Criteria
Definite Variant CJD
Neuropathologic examination of brain tissue is required to confirm a diagnosis of variant CJD. The following confirmatory features should be present.
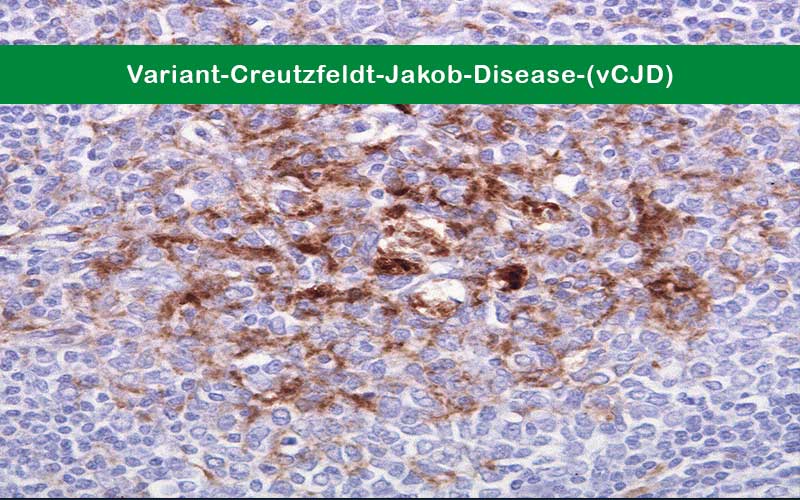
- Numerous widespread kuru-type amyloid plaques surrounded by vacuoles in both the cerebellum and cerebrum – florid plaques.
- Spongiform change and extensive prion protein deposition shown by immunohistochemistry throughout the cerebellum and cerebrum.
Suspected Variant CJD
- Current age or age at death <55 years (a brain autopsy is recommended, however, for all physician-diagnosed CJD cases).
- Psychiatric symptoms at illness onset and/or persistent painful sensory symptoms (frank pain and/or dysesthesia).
- Dementia, and development ≥4 months after illness onset of at least two of the following five neurologic signs: poor coordination, myoclonus, chorea, hyperreflexia, or visual signs. (If persistent painful sensory symptoms exist, ≥4 months delay in the development of the neurologic signs is not required).
- A normal or an abnormal EEG, but not the diagnostic EEG changes often seen in classic CJD.
- Duration of illness of over 6 months.
- Routine investigations of the patient do not suggest an alternative, non-CJD diagnosis.
- No history of receipt of cadaveric human pituitary growth hormone or a dura mater graft.
- No history of CJD in a first degree relative or prion protein gene mutation in the patient.


